SSMD: A semi-supervised approach for a robust identification of cell types and deconvolution of mouse transcriptomics data
Description
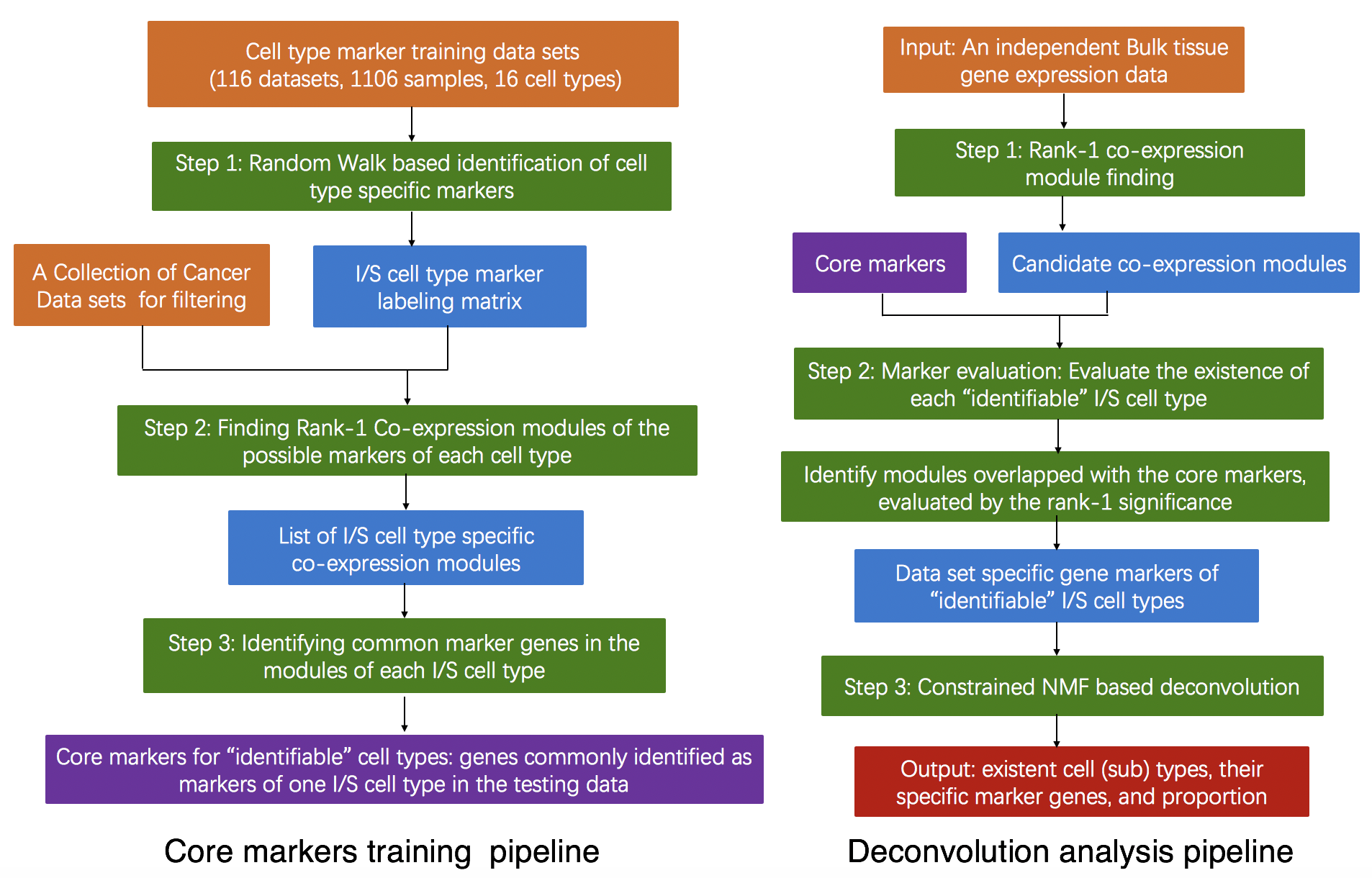
Multiple deconvolution methods have been developed for investigating the heterogeneous immune and stromal (I/S) cell types in human cancer tissue to estimate their relative abundances using transcriptomic data. However, there is a lack of a robust method and user-friendly software for mouse transcriptomic data deconvolution. Here, we developed a novel semi-supervised approach, namely SSMD, by (i) deriving potential I/S cell signature genes from a large collection of mouse data sets to form a marker labeling matrix; (ii) implementing a rank-1 sub matrix identification method to test the presence of I/S cell types and identify data set specific I/S cell markers; and (iii) utilizing a constrained non-negative matrix factorization (NMF) based framework to account for diversity of mouse models. The new method was validated on single cell RNA-seq simulated bulk tissue data and independent immuno-assay data. The method is applied to mouse prostate cancer data sets to infer the level of anti-cancer immune cell populations.
Installation
install.packages("devtools")
devtools::install_github("zy26/SSMD")
Usage
estimate.proportion <- function(data, lambda = lambda)
Arguments
datainput gene expression matrix. MGI gene symbol should be as their row namesparameterthreshold of mean correlation to define rank-1 co-expression module
Value
An object of class is also invisibly returned. This is a list containing the following components:
Stat_allstatistics for all rank-1 co-expression module.CT: cell type; mean: mean correlation inside the module;Core_overlap_number: Overlap number with core marker list;Core_overlap_rate: overlap rate with core marker list;BCV_rank: bcv rank of the first basemodule_keepmodules with the high overlap number with core marker list for each cell typeproportionestimated proportion for each cell type
Examples
#load your own gene expression data
load('example_bulk.RData')
estimate.proportion(data, lambda = 0.8)